Genomic Medicine
Talos shows automated genome reanalysis can deliver new diagnoses in weeks, not years
Talos reframes genome reanalysis as a continuous, automated program rather than a rare manual event. Across a prospective cohort of 4,735 undiagnosed patients, it yielded 241 new diagnoses (5.1% additional yield) within weeks of new evidence being published, while requiring analysts to review only one new variant per 200 patients per month.
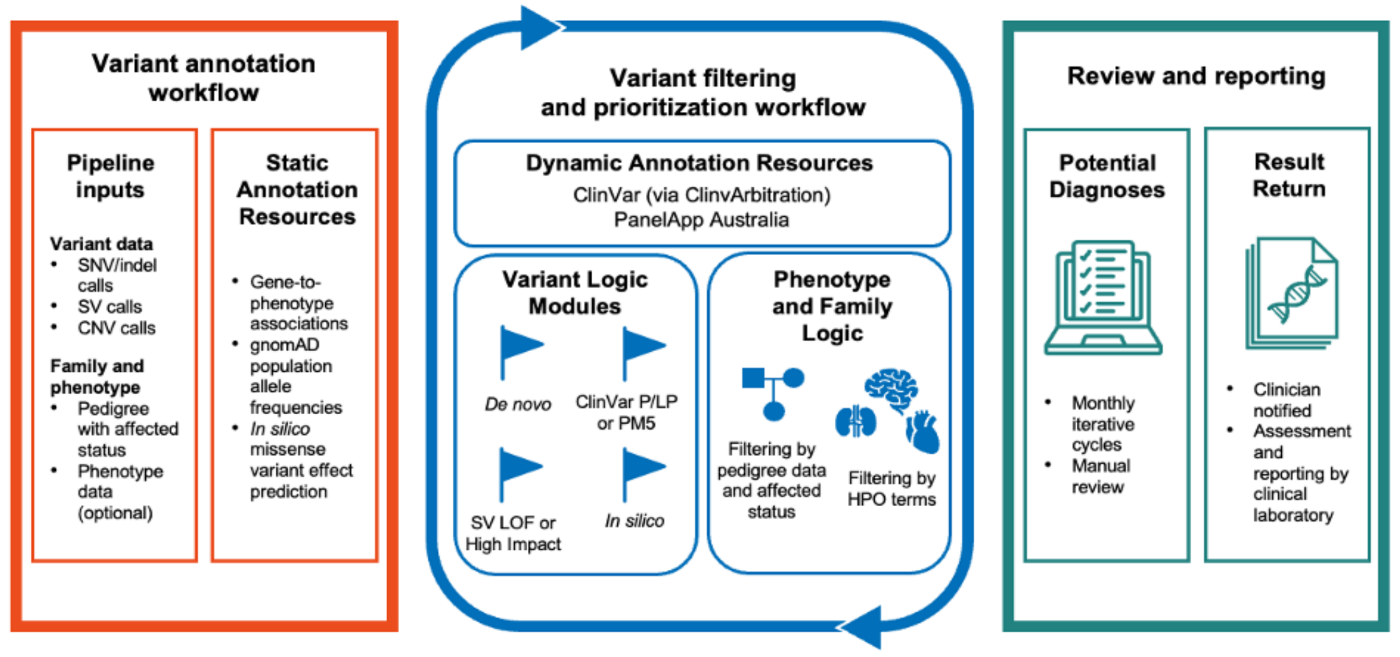
For most patients with rare genetic diseases, the first genomic test does not deliver a diagnosis, not because the answer is missing from the data, but because our understanding of the genome is still incomplete. New gene-disease associations and variant classifications come out every year. Stored sequencing data can be reexamined to find answers that were unknowable at the time of the original test. The problem has always been scale: reanalysis is mostly manual, dependent on motivated clinicians and scarce lab staff. As a result, most genomes are never revisited.
An international team from the Centre for Population Genomics, Australian Genomics, the Broad Institute, and Microsoft has now published a solution. Talos, an open-source tool for automated, iterative reanalysis of genomic data, shows that systematic reanalysis can be feasible and valuable at scale. The tool is deliberately conservative. It returns a small set of high-confidence variants instead of a long ranked list. It focuses clinicians on variants whose supporting evidence has changed since the last analysis cycle.
Design philosophy: specificity over recall
Talos reinterprets a patient's existing variant calls against two continuously updated public resources: PanelApp Australia for gene-disease relationships and ClinVar for variant-level pathogenicity. It then runs a prioritization algorithm designed to surface variants most likely to meet ACMG/AMP criteria for clinical reporting. The pipeline uses family structure, mode of inheritance, and de novo status, along with phenotype data when available, to narrow the candidate set.
Two design choices set Talos apart from other tools. First, it is optimized for a low false-positive rate. Across a validation set of nearly 1,100 patients, it recovered 90% of in-scope diagnoses while flagging only 1.3 candidate variants per patient for expert review. Second, on repeat runs, Talos returns only variants whose supporting evidence has changed since the previous cycle, so clinicians see genuinely new findings.
Validation against expert manual analysis
The team benchmarked Talos against two independent cohorts that had already undergone careful manual analysis: the Australian Acute Care Genomics cohort of critically ill infants and children, and the U.S.-based Rare Genomes Project cohort of families with prior uninformative testing. On the ACG trios, Talos recovered 90% of in-scope diagnoses at a median of 1.3 candidate variants per family. The diagnoses it missed were mostly a consequence of its conservative strategy, recessive variants lacking ClinVar support that human analysts had classified using trans configuration or functional studies.
Crucially, Talos held the same operating point on the RGP cohort, which included probands ranging up to 82 years of age. On those trios, it recovered 87% of in-scope diagnoses at a median of 1.3 candidate variants per trio, showing it generalizes across cohorts.
In head-to-head benchmarking against Exomiser, a widely used prioritization tool, Talos matched its overall sensitivity for small variants but operated at a very different point on the precision-recall curve. Exomiser ranks and returns a broad list. Talos returns a short, highly specific one. In paired comparison, the two tools were statistically indistinguishable when all of Exomiser's ranked variants were reviewed, but Talos came out significantly ahead once review was limited to a realistic budget: the top five (p = 0.017) or top one (p < 0.0001) ranked variants. Notably, the two tools surfaced different variants, meaning they are complementary and should be used together in diagnostic workflows.
Deployment at scale
Talos was then deployed on a tested-but-undiagnosed cohort of 4,735 individuals drawn from Australian Genomics research studies and a single diagnostic laboratory. Most patients were singletons with neurodevelopmental, cardiac, renal, or neurological indications.
Talos produced 241 new diagnoses in 238 individuals, a 5.1% additional yield, with every single likely-causative variant subsequently confirmed as pathogenic or likely pathogenic by accredited labs. The sources of those diagnoses illustrate why reanalysis is such a powerful approach:
- 32% came from new gene-disease relationships discovered since the original test
- 22% came from new variant-level evidence (reclassifications)
- 45% came from improved filtering and analysis, including variant types such as CNVs and structural variants not examined originally, phenotype filters that had been set too narrowly, and other sources
Yield was consistent across clinical areas (roughly 5 to 6% for neurodevelopmental, cardiac, and renal indications), but the reasons differed. New gene associations and CNVs dominated neurodevelopmental diagnoses, while variant reclassification drove most cardiac ones. Genome data outperformed exome (6.1% versus 4.8%), partly by reaching non-coding diagnoses such as RNU4-2 and a deep-intronic MRPL39 variant. A recurring theme was the lag in conventional knowledge bases: 59% of the new gene-disease diagnoses were not yet curated in OMIM at the time of reanalysis, highlighting the value of using a rapidly updated resource like PanelApp Australia.
Iterative reanalysis: from one-off event to continuous program
The team then ran Talos for 29 monthly iterative cycles. Most diagnoses (92%) came on a cohort's first pass, but the iterative design proved its value on two fronts. First, it showed the scalability of ongoing reanalysis. Because later cycles return only newly actionable evidence, they surfaced an average of just one variant per 200 cases over the program. Second, it showed how quickly the system could move from scientific discovery to diagnosis. On average, just 32 days passed between new knowledge appearing in a public database and a patient receiving a diagnosis. The fastest case turned around in a single day.
The pipeline is cheap enough to run continuously: annotating 1,000 genomes cost about $11, and a monthly reanalysis pass ran for a few cents per cohort.
Looking ahead
Talos reframes genomic reanalysis from a rare, labor-intensive event into a continuous, automated program that can keep pace with the science. By optimizing for specificity, it respects the real bottleneck of expert reviewer time. By drawing on openly shared, frequently updated resources, it turns the global community's accumulating knowledge into diagnoses for individual patients, often within weeks.
The tool is open source and straightforward to deploy in cloud environments like Azure. The team behind Talos sees it as a foundational capability and is looking forward to incorporating more advanced AI models for understanding and predicting the consequences of genetic variation into the reanalysis of unsolved rare disease cases.